






新しい医薬品ができるまで
- HOME
- 薬の基礎知識を得るには?
- 新しい医薬品ができるまで
1. 基礎研究について
医薬品になる可能性のある物質(候補物質)を探すための研究です。植物や動物等の天然物から物質を取り出したり、化学的に合成したりします。抗体等のタンパク質やインスリン等のペプチドについては、バイオテクノロジー(遺伝子組換え技術等)を利用して作ります。
候補物質について細胞や動物等を使った生物学的試験等で効果や大まかな毒性等を調べます。また、候補物質が生体で働きやすいように、構造を改変したりします。
2.非臨床試験について
開発すると決めた候補物質について、以下の試験をします。これらの成績により、人でも薬効(有効性)が得られ、かつ安全性に問題ないかどうかを推測します。そして、人での試験をするかどうかを判断します。このため、これらの試験成績は信頼がおけるものである必要がありますので、試験の実施にあたり法規で定められた基準を守る必要があります。
a) 品質に関する試験(非臨床試験とは別に分類されることもあります。)
一定の効き目や安全性を担保するためには、一定の品質の物質が必要です。
一定の品質の規格とその試験方法(①)やどのくらい品質が安定か(②)を調べる試験です。候補物質の品質に関する試験には、以下があります。
①製造方法並びに規格及び試験方法に関する試験(構造決定及び物理化学的性質等、製造方法、規格及び試験方法等)
②安定性に関する試験(長期保存試験、過酷試験、加速試験)
b) 薬理作用に関する試験
いろいろな実験動物や細胞等を用いてどの位の用量で効果があるのか(①)、また、そのように効果が現れるのか(①)、さらに、効果が現れる用量で中枢神経、心血管系や呼吸器系等に影響がでないか等(②、③)を調べる試験です。薬理作用に関する試験には、以下があります。
①効力を裏付ける試験(薬効・薬理試験)
②副次的薬理試験・安全性薬理試験
③その他の薬理試験
c) 吸収、分布、代謝及び排泄(体内動態)に関する試験
動物(薬効が現れる動物)を用いて、候補物質が生体でどのように吸収され、分布し、代謝(解毒)され、そして排泄されるか(体内動態)を明らかにします。
以下の試験があります。
①吸収、②分布、③代謝、④排泄 等
d) 毒性性に関する試験
動物(薬効が現れる動物)を用いて、薬効が現れる用量より多い用量での毒性を調べます。通常は、毒性が現れる用量、薬効が現れる用量に近い用量で毒性が現れないこと、命にかかわるような毒性が現れないこと等を確かめます。また、どのような毒性があるのか、どの程度の用量から毒性の現れるのか(最少毒性発現用量)等を明らかにして、人に投与する場合の用量の安全性の情報とします。以下の試験があります。
①単回投与毒性、②反復投与毒性、③遺伝毒性、④生殖発生毒性、⑤局所刺激性、
⑥トキシコキネティック試験、⑦その他の毒性 等
人に投与する前に必要な安全性に関する情報を得るための重要な試験ですので、信頼のおける情報を得る必要があります。このため、その実施は、医薬品の安全性に関する非臨床試験の実施の基準(GLP)に適合した試験施設での実施が必要です。「b)薬理作用に関する試験」のうち、安全性薬理試験の実施もGLP適合施設での実施が必要です。
3.臨床試験について
非臨床試験で薬効が十分であり、毒性(安全性)に問題がないことを確認した上で、人での薬効(有効性)と安全性を確認します。人を対象とした試験を臨床試験といいますが、その中で医薬品の承認申請書に添付する資料の収集のための試験の実施を「治験」といいます(医薬品医療機器等法第2条第16項)。
治験はGCP(医薬品の臨床試験の実施の基準)を遵守して、倫理的かつ科学的に実施することが求められています。
通常は以下の3つの(①第Ⅰ相試験、②第Ⅱ相試験、③第Ⅲ相試験)について、安全性に十分注意しながら、①→②→③の順に段階的に行います。そして、それぞれの段階での結果を踏まえて、次の段階に進めるかどうかを検討しながら慎重に試験をすすめます。
①第Ⅰ相試験:
人での吸収・排泄等の薬物動態と安全性を調べます。
通常は少数の健康成人に安全な用量から薬効が現れる用量まで投与し、採血等をして候補物質の血液中の濃度等を調べ、また、安全性に問題がないか(副作用等が起きないか)を調べます。
②第Ⅱ相試験:
薬効(有効性)が期待できる安全な用法・用量(例えば、1日2回、それぞれ〇mg等)を探索し、設定します。
少数の対象疾患の患者に、第Ⅰ相試験で安全性が確認された範囲の用量の中で薬効が期待できる用量を複数(通常3種類程度)投与します。そして、効果(事例:脂質異常症の治療薬の場合は、血中のコレステロールの値の変化)を見て、どの用量・用量が良いかを、その用法・用量での安全性も勘案して検討し、最適な用法・用量を設定します。
③第Ⅲ相試験:
第Ⅱ相試験で設定された用法・用量で臨床試験を行い、有効性及び安全性を検証します。
多数の対象疾患の患者さんを対象に臨床試験を行い、設定された用法・用量を一定期間投与した場合に効果が得られるか、また、安全性に問題がないかを調べます。
第Ⅲ相試験では、通常は対照群(プラセボを服用する群)と候補物質を服用する群(被験薬群)の効果等を比較します。その際には、患者さんがどちらの群に入っているのかは、患者さんも医師も知りません(盲検化といいます)。また、疾病の程度や性別や年齢が偏らないように無作為(ランダム)に両群に患者さんを振り分けて、両群の結果を比較します。このような試験を、無作為化(ランダム化)プラセボ対照二重盲検比較試験といいます。この方法でプラセボ群と被験薬群を比較することにより、自然治癒力等により疾病が治癒したのではなく、候補物質に本当に効果があったかどうかを明らかにすることができます。
4.承認申請 (製造販売承認申請)
医薬品の候補物質を医薬品として市販する(製造販売する)には、その物質の品質、有効性及び安全性に関する資料(承認申請資料)を添えて厚生労働大臣に製造販売承認申請し、審査を受けて、厚生労働大臣の承認をうけなければなりません(医薬品医療機器等第14条)。
この申請は、医薬品の製造販売業者(製造販売業の許可を得ている製薬企業)のみしか行えません。製造販売業者は、厚生労働大臣に上記「2.非臨床試験」と上記「3.臨床試験(治験)」の成績を纏めた資料を添付した申請書を添えて提出します
また、医薬品の添付文書も添付して提出します。
申請書に添付しなければならない資料は、以下の通りです。
医薬品の承認申請の際に添付すべき資料
(医薬品医療機器等法施行規則第40条第1項より第1号を抜粋)
医薬品(体外診断薬を除く)について次に掲げる資料
イ 起原又は発見の経緯及び外国における使用状況に関する資料
[1.起原又は発見の経緯、2.外国における使用状況、3.特性及び他の医薬品との比較検討等]
ロ 製造方法並びに規格及び試験方法等に関する資料
[1.構造決定、2.物理化学的性質等、3.規格及び試験方法]
ハ 安定性に関する資料
[1.長期保存試験、2.過酷試験、3.加速試験]
ニ 薬理作用に関する資料
[1.効力を裏付ける試験、2.副次的薬理・安全性薬理、3.その他]
ホ 吸収、分布、代謝及び排泄に関する資料
[1.吸収、2.分布、3.代謝、4.排泄、5.生物学的同等性、6.その他の薬物動態]
ヘ 急性毒性、亜急性毒性、慢性毒性、催奇形性その他の毒性に関する資料
[1.単回投与毒性(急性)、2.反復投与毒性(亜急性、慢性)、3.遺伝毒性、4.がん原性、5.生殖発生毒性、6.局所刺激性、7.その他の毒性(依存性試験、抗原性試験、皮膚(光)感作性試験等)]
ト 臨床試験等に関する資料
[臨床試験成績]
チ 法第52条第1項に規定する添付文書等記載事項に関する資料
[ ] 内は平成26年11月21日薬食審査発第1121第12号号 厚生労働省医薬食品局審査管理課長通知「医薬品の承認申請に際し留意すべき事項について」より抜粋して追記
5.承認審査
医薬品の製造販売業者から承認申請された品目について、その承認申請資料に基いて、(独)医薬品医療機器総合機構(PMDA)において審査等が行われます。
審査は、以下の点等について行われます。
①予定されている効能・効果、用法・用量での安全性及び有効性が示されているか、さらにベネフィットがリスクを上まわるか。
②予定されている添付文書等の表示が適切か。
③製造所において、製造管理及び品質を維持するための品質管理方法が、厚生労働省令で定める基準に適合しているか。
審査等の流れは以下の図を参照して下さい。図の申請者は「医薬品の製造販売業者」です。
承認申請資料は科学的で倫理的でかつ信頼できるものでなければなりませんので、厚生労働省令の定める基準(GCP、GLP等)に従って収集され、かつ、作成されたものであることが要求されています(同法第14条第3項)。これを確認するための調査を信頼性調査といいます。
審査は、提出された承認申請資料(品質、薬理、薬物動態、毒性及び臨床試験に関する資料)について、医学、薬学及び生物統計学等の専門家からなるチームが行います。審査及び信頼性調査の結果を、審査報告書に纏めます。
さらに、申請された品目が厚生労働大臣の許可を受けた製造所において製造管理の基準(GMP)に基づき製造されているかどうかも調査されます(GMP調査)。
さらに、申請された品目が厚生労働大臣の許可を受けた製造所において製造管理の基準(GMP)に基づき製造されているかどうかも調査されます(GMP調査)。
医薬品の承認審査業務のフローチャート
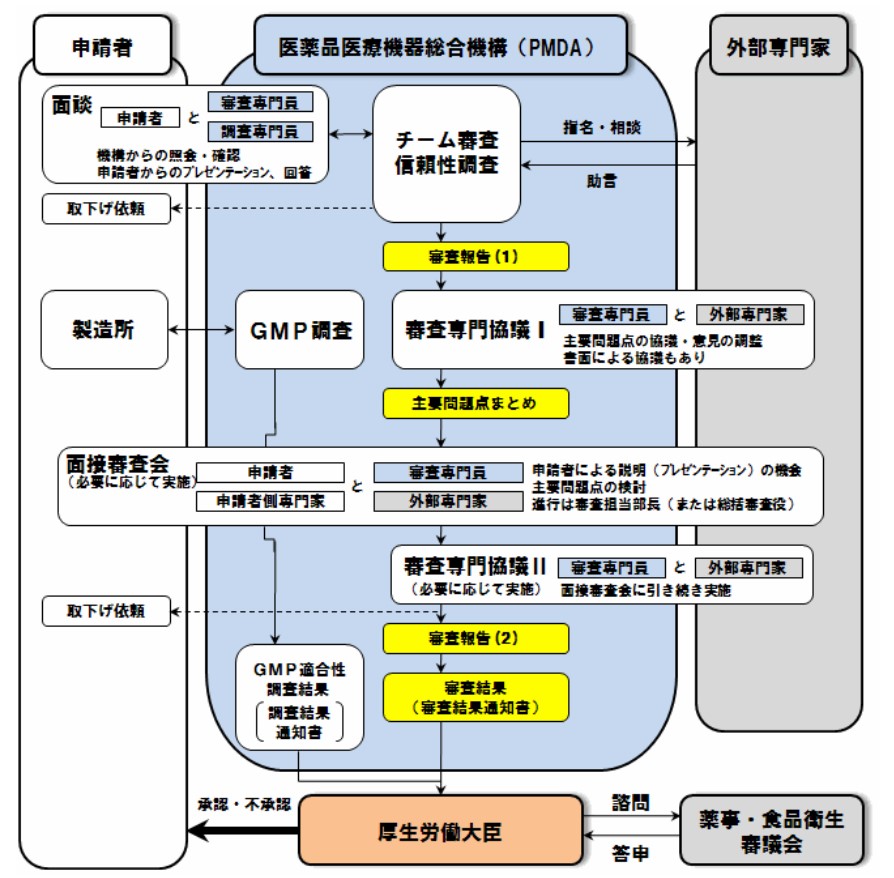
6.承認(製造販売承認)
PMDAによる審査の結果(審査報告書)をうけて、厚生労働大臣は薬事・食品衛生審議会の意見を聴いて(諮問して)、同審議会の回答(答申)を受けて、製造販売承認の可否を判断します。
参考資料等:医薬品医療機器等法
https://www.nibiohn.go.jp/nibio/guide/page1.html
医薬品開発学入門 じほう社
2019-20年版 薬事関係法規・制度 解説 薬事日報社
(更新日 2021年8月6日)